Summary or TL;DR – There are many different aproaches available to making a DNA library – which simply is a collection of different DNA sequences containing variation in one or more positions. Each library generation method deals differently with a number of parameters, such as location of the mutations and the number of variants generated. In most circumstances, it is possible for multiple methods to deliver the same library at different cost, or different libraries at the same cost. The challenge then becomes in choosing a suitable method that delivers a suitable library for the application at hand.

Use of metaphors: To make some of the concepts more accessible and to emphasise particular differences between approaches, I use a LEGO set metaphor here. The original kit is your starting point or wild-type protein and evolution here is focusing on changing some of the bricks in the set so that you get a different toy out of your starting set. Libraries are the parts you can order from the LEGO shop with the methods being how you can order those parts. The cost in this shop is set by the number of orders you place.
One of the key and most fundamental considerations in directed evolution is the starting library. For instance, the number of variants within a library should be compatible with the approach used in selection to make sampling meaningful: a library that exceeds 1e10 (1 x 1010) variants is not effectively sampled if a selection platform is limited in its potential to screen 1e5 variants at a time. In our LEGO analogy, this would be the equivalent of only having your own two hands to test the 10,000 spare parts you ordered.
However, library size (in relation to the selection platform) is only one aspect of what is generally described as library quality. High quality libraries target mutations to the region of the DNA being selected (e.g. to the gene not the plasmid carrying the target gene for evolution) and they minimise the number of inactive variants (which would therefore not contribute to selection). It also minimises the frequency of the starting gene (which, presumably, does not have the desired function) and it minimises biases from the genetic code sampling (e.g. there are six codons for leucine but only one for methionine). The latter is important because it directly contributes to library size. In our LEGO analogy, this would be the equivalent of being able to order a single copy of all parts that match the space available, while excluding the starting piece you are trying to replace. Clearly, the ideal scenario in the LEGO situation would be to order only the single piece you want and that is the goal of rational protein design – a topic for a different blog.
In practice, it is not always technically possible or economically feasible to achieve perfect libraries and some compromise is required. The commonest and most economical approach towards generating libraries of contiguous mutations is to introduce degeneracies in your oligos. This means that rather than having a single oligonucleotide, you have a mixture of oligonucleotides that are stochastically incorporated by PCR to synthesise the library. For instance, a degenerate oligo such as NTT (degeneracies are represented by standard letters defined by IUPAC conventions – see here) would include a mixture of TTT, CTT, ATT and GTT that would result in F, L, I or V being incorporated in the resulting protein. This would be the equivalent of being able to specify that your block must be thin and four pegs by two long when ordering: you will have blocks of different colours that match your description.

For proteins, in situtations where all amino acids can be considered as possible substitutions, a degeneracy NNK is used, resulting in 32 possibilities to cover the 20 amino acids and 1 STOP codon. This would be equivalent to ordering every 2-peg-wide piece available – different colours, different lengths, different thicknesses. Targetting multiple positions with NNK rapidly increases the size of the library, with a library of 8 NNK containing 1.1e12 DNA variants (32^8). Still, because of the redundancy of the genetic code, the 1.1e12 DNA variants sample only 3.8e10 protein variants (21^8) – i.e. nearly 96% of the library is redundant. Worse still, only about 68% of those 3.8e10 proteins will not include a STOP codon (which in NNK is encoded as UAG). This would therefore be the equivalent of ordering every piece in the shop (as a single order) and you get 6 blue pieces for each yellow one. You can now build any toy, but you have to sort through a huge box.
Because the NNK approach has been very successful in both academia and industry, it can be misleading to think that it is the best or the only approach available for the synthesis of DNA libraries. If a single residue is being targeted for mutation, combinations of partially degenerate oligos have been shown to be highly effective – see here. Still, the limitations of the degenerate oligos approach begin to be felt when, supported by functional understanding, the design of the library indicates that only a subset of the possible 20 amino acids needs to be sampled, or that some amino acids should be more frequently sampled than others. In other words, it is great that you can buy every piece from the LEGO shop but you may not have the time to test them all. It becomes more efficient to be able to describe the particular pieces you need.
For instance, if a flexible linker is being optimised, it is possible that the design would favour small, flexible and polar residues for incorporation. In this case, a degeneracy such as RST would allow coverage of ACT, GCT, AGT and GGT coding respectively for T, A, S and G, which are the small, flexible and polar amino acids. It would also be possible to adjust the degree of degeneracy during oligo synthesis so, for instance using 75% G and 25% A in the first R position, would result in a 3 (A and G) to 1 (T and S).
Nonetheless, design may require a more specific choice of amino acids in a given position. Let’s say the requirement is for a sulphur-containing amino acid, i.e. C or M. In such a situation, oligonucleotide degeneracy to provide the 2 desired amino acids (WKS) bring with it two ‘irrelevant’ amino acids, F and R. On the surface, this may not seem to be a problem, particularly for in vitro selection platforms that can cope with 1e14 variants in a single selection round.
Consider however a simple library where 8 positions are being targeted for the screening of C or M. The ideal library represented only by the required amino acids contains 2^8 (or 256) variants. Generating such a library with degenerate primers creates 4^8 (65,536) variants – i.e. the desired library would be only 0.3% of the total library. Other examples can be readily explored and whenever degeneracy incorporates non-desired variation, the quality of the library is strongly affected. I will in future edits add here a code in Julia to better allow these comparisons, but there are also online tools that can be used to optimise degenerate oligos – see here.
In theory, it is possible to generate each needed variant by carrying out individual PCRs with primer combinations being used to deliver the exact mutants needed – the equivalent of being able to order from the LEGO shop the parts by their part number. For the (C/M)x8 library above, a 32-primer set would suffice to generate all possible combinations. Primer mixtures could be made and the library assembled in a single PCR – with the desired library being 100% of the total library. Still, as the library complexity increases, so do the number of required primers: a design for 4 amino acids sampled in 8 residues could require 512 primers, with an estimated cost of over EUR2,000. This would be the equivalent of having to place 512 orders, each for the particular brick you need.
Note: Because the diversity can be split between two primers (assuming these are being introduced by PCR), primer design can be optimised with diversity for the first four residues in one primer and diversity for the last four residues in the second primer. Assuming the 4 residues cannot be encoded with degeneracies, 256 (4x4x4x4) variants of each primer would be required to generate the library.
Nevertheless, other platforms for DNA library synthesis also exist: additive synthesis platforms where individual codons are added to generate highly customisable libraries – see here, here and here. For the (C/M)x8 example above, such methodologies would require 8 synthesis cycles, each cycle with two individual building blocks. If greater diversity of mutation is required (as in the example above), additive synthesis strategies are more scalable and remain cost-effective (e.g. for the 4 amino acid, 8 residue library, 4 building blocks repeated 8 cycles would generate the library). This would be the equivalent of you being able to send a photo of your kit, highlighting the piece you want to substitute and the shop being able to send you one of each piece that would fit that space. Even if the results are the same as ordering multiple parts, this approach always results in a single order from your favourite LEGO shop.
The advantages of additive-synthesis-based methods over PCR-based methods become only more pronounced as library complexity increases, particularly if library designs incorporate sampling biases for certain amino acids at certain positions, if library (or building block) length is variable and if library size is an important variable being considered.
A more extreme library would be ABCDEFGH, where each letter represents a single amino acid. If libraries of varying length from that motif are part of the design (the reasons for that style of library design and for that approach not being commonly adopted are a whole separate blog), degeneracy or multiple primers need to be considered. Multiple primers would require 257 primers to cover all the possible combinations of 8, 7, 6 and so on residues in such a complex library. Degeneracy is possible but, as detailed above for single-length sequences, it will invariably compromise library quality: sampling ‘undesired’ mutations in a rapidly expanding library.
In InDel assembly (which encompasses both the assembly and the subsequent analysis) we have developed a platform of additive library synthesis that, by not selecting for 100% incorporation of the building blocks, creates libraries that vary both in length and composition. For synthesising protein libraries, like other additive synthesis methodologies, the building blocks are simple and few: 20 building blocks are all that are needed for any assembly based on single codon cycles.
Shorter libraries sample the whole of the sequence space provided by the building blocks used, larger libraries converge towards the assembly program. In theory, the more restricted the use of the building blocks is, the more the library looks like the result of natural insertion or deletion events – hence the name of the platform. So, for an Indel library of the form ABC, to obtain the full-length desired sequence, an assembly program like AABBCC would be used. Based on coupling efficiencies of 20% per cycle, 3% of the final assembly would be the desired ABC design. Variants of 0 (a full deletion) and 6 (the complete assembly program) amino acids would be generated as well as libraries of 1, 2, 3, 4 and 5 amino acids. Such a library of libraries would be synthesised in little over a day.
By PCR, it depends immensely on whether ABC can be coded under a single degeneracy. If they can, with 7 primers it is possible to generate a similar library with longer sequences better represented – assuming, of course, that the library design would consider a variant such as BBBBBB relevant. In practice, many in the field believe that as you move away from the starting sequence, you are less likely to isolate a functional variant – see here. A multi-primer setup could deliver a more similar library, and it should come to approximately 78 primers.
On the other hand, if A, B and C represent mixtures of codons (including also different ratios of codons) or building blocks of different lengths (e.g. secondary structure elements), then PCR-based methods cannot compete with additive synthesis approaches – the quality of the libraries will (in all likelihood) simply be too low for most selection techniques.
For developing and validating InDel, we focused on the omega loop in beta-lactamase. The enzyme, responsible for ampicillin (an antibiotic) resistance in bacteria, is a common well-characterised and easily assayable enzyme in microbiology, and a model target for directed evolution. The omega loop is equally well-characterised for its role in substrate specificity and it had been previously shown to be functional at different lengths. Together, this made it a suitable target to develop and validate the technology (both synthesis and subsequent analysis). The wild-type omega loop is DRWEPEL and we knew from the outset that an engineered loop DRYYGEL – see here – would give us a measurable different antibiotic resistance profile.
Our library was assembled as shown in the figure below:
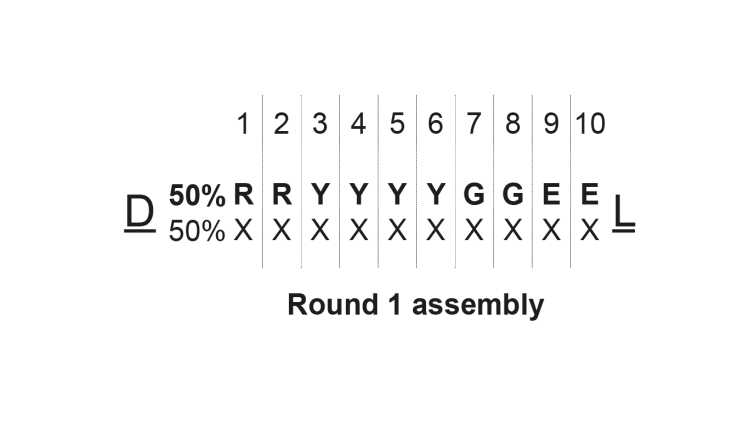
Each cycle bringing 50% of our target sequence and 50% of a mixture of all 20 building blocks available. The goal was to generate noisy data to also demonstrate the power of the analysis platform.
The output library was expected to look like:

The InDel library shown above is skewed towards shorter sequences than the program as it would be expected from the incomplete incorporations per cycle. In addition, it is heavily skewed towards the immediate sequence neighbourhood of the target sequence (but without some of the biases that would emerge in error-prone PCR). Longer lengths start to increasingly resemble the assembly program (RRYYYYGGEE). In this simulation, the intended target (RYYGE) is present at 0.01% . That would mean that in a usual 1e8 library, the target sequence would be expected to be present approximately 10,000 times.
PCR can be used to assemble a family of libraries of the same length, but they will almost invariably be of very distinct profile. For instance, if a pure (NNK)x6 strategy is used, 1.1e9 DNA variants are generated (representing 6.4e7 proteins) with the intended target sequence RYYGE comprising 0.00002% of the total sequence with an expected 17 copies in a 1e8 library. Such infrequent occurrence is already a problem even before libraries of other lengths are generated and added to the complex library. And here again the LEGO metaphor is useful, because selection is picking a random sample of your bricks and seeing if they fit your kit, if they don’t, the bricks go back in the bag and you try again. If your box has 1e8 pieces finding one of your target pieces is a lot easier if there are 10,000 of them in the box, rather than 17.
On the other hand, other PCR designs are possible, for example, where instead of NNK a partial degeneracy is used. A 70%C10%GAT 70%G10%ACT K in the first residue would create a biased library towards the incorporation of R (it would lead to 52% of R incorporation at that position with other amino acids represented between 0.5% and 7.5%). That approach would lead to a library biased towards the target sequence with RYYGE representing 0.1% of the population. Other biases would remain (and it still would look different from the InDel generated library) and in future updates I will provide a more detailed comparison for this point, including also a comparison to error-prone PCR.
Such customised mixing has to be done manually when chemically synthesising the primers, leading to significant increases in cost – e.g. the synthesis of the 5-codons needed to cover RYYGE at IDT come to EUR 386.90 at RRP. To obtain the different lengths, multiple primers (a total of 7 to cover libraries between 1 to 6 amino acids) would be required and the total cost of generating such library would exceed EUR1,500. We estimate an InDel library to cost approximately EUR150.
Nonetheless, the precision of assembling a starting library to a given design hinges on whether the information used in design is correct (and not limited by database mis-annotations or incomplete knowledge of the target enzyme). In suboptimal cases, sampling beyond design parameters can yield results if your patience, luck and funding are good enough.
Still, the power of InDel is not in the synthesis of loop libraries, even if we did demonstrate its potential in that simple system. Its power is in the freedom to combine building blocks of any length allowing the synthesis of complex modular nucleic acid sequences (e.g. metabolic pathways and DNA nanostructures) as well as in the combinatorial assembly of protein genes (e.g. building proteins from secondary structure elements). Moreover, the analysis pipeline established with InDel assembly, allows the rapid processing of NGS data from selection and the isolation of functional peaks. Back to our LEGO analogy, this would be the equivalent of you being able to send a photo of your kit, highlighting the piece you want to substitute and the shop being able to send you one of each piece that would fit that space, including combinations of pieces to give you the fit. Coupled with the analysis pipeline, what you have is an incredibly fast way to find out which pieces fit best. It’s like having a LEGO shop that can read your mind. Now that is one unique LEGO shop.
Clarification: I am not the first to consider the importance of the quality of DNA libraries and updated versions of this post will include at least some of the relevant literature that tackles the issues raised here. Ultimately, this is a mathematical problem that is part of the routine for scientists working in directed evolution and that can have huge impact on a project’s success.
